Medical Device Regulation EU and CE Mark
Medical Device Regulation EU or CE Mark on device
labels and pack proves that device(s) meet the current EU medical device regulation or medical device manufacturer’s claim that a product meets the General Safety requirements of European Regulation. The Regulation outline the safety and performance requirements for medical devices in the European Union (EU).CE Mark for Medical Devices is a legal requirement to sell a device in European Union countries.
Before affixing medical device CE Marking, the device must follow the definition and be classified correctly following the classification rules laid down in annexes VIII of the EU MDR 2017/745. Medical devices are divided into four risk classes: I (Im, Is), IIa, IIb and III. Class, I medical devices are lower risk devices, while class III medical devise are the highest risk devices. The manufacturer of a class I device is responsible for the self-declaration of the MDR CE marking process. A notified body must certify medical devices belonging to higher risk classes.
CE Mark for Medical Devices on labels and packing proves that device(s) meet the current EU medical device regulation or medical device manufacturer’s claim that a product meets the General Safety requirements of European Regulation.
The Regulation outline the safety and performance requirements for medical devices in the European Union (EU).CE Mark for Medical Devices is a legal requirement to sell a device in European Union countries.
Before affixing Medical Device CE Marking, the device must follow the definition and be classified correctly following the classification rules laid down in annexes VIII of the EU MDR 2017/745. Medical devices are divided into four risk classes: I (Im, Is), IIa, IIb and III. Class, I medical devices are lower risk devices, while class III medical devise are the highest risk devices.
The manufacturer of a class I device is responsible for the self-declaration of the MDR CE marking process. A notified body must certify medical devices belonging to higher risk classes.
The Medical Device Regulation EU is a new set of regulations that govern medical devices’ production and distribution in Europe. Compliance with the regulation is mandatory for medical device companies that want to sell their products in the European marketplace.
The EU MDR regulation is intended to improve the safety and performance of medical devices in Europe and, as such, intends to provide a high level of protection for the health of patients and users of these medical devices.
Technical File as per Annex II of EU MDR 2017/745 is a technical document prepared by the manufacturer in a clear, well-organized, and unambiguous manner to demonstrate the general safety and performance of the device in question. The MDR Technical File for Medical Device of Higher class require notified body intervention or submitted to Competent Authority for review and approval.
This technical file should be preferably made in English or an official language of an EU Member state. In addition, for the Class I Medical device, a technical file must be prepared for self-declaration of CE mark over labels.
It must be available on request for the whole life cycle of the medical device (5 years for low risk and 16 years for high-risk devices) until and unless there is a change in device design. Non-EU manufacturers shall share the technical file with the authorized representative in the EU.
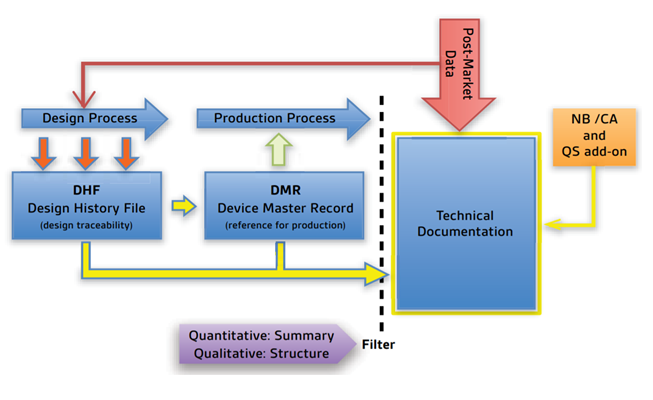
The technical documentation is prepared to demonstrate the conformity to the Medical device’s general safety and performance requirements as per European Union Medical Device Regulation 2017/745. It includes the device design and development, clinical evaluation and performance validation, conformity assessment and regulatory status within target markets.
Furthermore, the MDR now requires a closed-loop process, implemented with data from the post-market use of the device (PMS), to ensure that early warnings are captured, and the ‘General Safety and Performance Requirements (GSPRs) are continuously fulfilled that the benefits for the patient always outweigh the risks.
- Identify the device for CE Marking
- Appoint experienced consultants
- Decide the directive, class and route of CE Marking
- Implement ISO 13485
- Conduct Risk analysis
- Appoint European Authorized Representative
- Perform safety and functional testing
- Perform clinical evaluation
- Perform critical process validations
- Prepare Technical file / Design dossier
- Appoint a Notified Body
- Submit technical file for Notified Body Review
- Answer to Notified Body Concerns and update the technical file
- Notified Body site audit
Affix CE Mark on your product.
